Mass Spectrometers for Proteomics
Orbitrap Astral Zoom Mass Spectrometer

The Astral Zoom MS is our lab’s newest MS instrument, which combines fast acquisition speeds of up to 270 Hz with high resolution and sensitivity. The Astral Zoom supports advanced acquisition strategies such as high-resolution DIA and DDA, making it ideal for accurate, high-throughput proteomics (up to 300 samples per day), with capabilities for both label-free and TMT-based quantitation, as well as more specialised applications.
Orbitrap Eclipse Tribrid Mass Spectrometer

The Orbitrap Eclipse Tribrid MS allows our researchers to identify and quantitation proteins as well as post-translational modifications using electron-transfer dissociation (ETD). It incorporates the brightest ion source, a segmented quadrupole mass filter with improved selectivity and ion transmission, Advanced Vacuum Technology for improved ion transmission to the Orbitrap mass analyzer and higher-capacity ETD fragmentation. Combined, these hardware improvements enable the new tribrid instrument to excel in the most challenging applications, including analysis of low level PTMs (phosphorylation, glycosylation), multiplexed relative quantitation using isobaric tags (TMT) and more accurate Real Time Search (RTS) Synchronous Precursor Selection (SPS) MS3 method, protein-protein interaction using MS-cleavable crosslinking reagents, as well as Data-Independent Acquisition (DIA).
Automated Sample Preparation
Seer SP200

We are the first lab in the UK to set up the Seer Proteograph ONE low abundant plasma protein enrichment workflow with automated sample preparation on SP200 robot. In addition to Seer’s proprietary nanoparticles for low abundant protein enrichment, the automated workflow allows us to reproducibly process biological samples in a high throughput manner within 6 hours, with minimal manual handling. Unlike immuno-based, targeted methods, which are limited to predefined panels, mass spectrometry combined with protein corona enrichment enables the unbiased detection of thousands of proteins in plasma, including low-abundance species. The dedicated Proteograph Analysis Suite using cloud computing allow us to run data analysis and QC in a high speed and reproducible manner, enabling powerful biological insights.
10.1016/j.jprot.2025.105519
10.1101/2025.08.12.25333522
Bravo AssayMAP

The Agilent Bravo AssayMAP is our current workhorse for sample preparation and peptide clean up. With the build-in workflows and different types of cartridges, we can process samples in 96-well plate format with minimum handling and generate reproducible peptide samples ready for LC-MS/MS.
Mass Spectrometry Workflows
Label-free Proteomics
We apply untargeted proteomics to broadly profile proteins and their modifications in complex biological samples. Using both data-dependent acquisition (DDA) and data-independent acquisition (DIA), we can identify and quantify thousands of proteins in a single experiment. These methods also allow us to detect important post-translational modifications (PTMs), such as phosphorylation or glycosylation, which play key roles in regulating protein function. Together, DDA and DIA give us comprehensive and reproducible insights into protein expression, regulation, and pathways in health and disease. Proteome Discoverer with Mascot algorithm are used for analyzing DDA data and Spectronaut for DIA data.
Tandem Mass Tag (TMT)-based Protein Quantitation
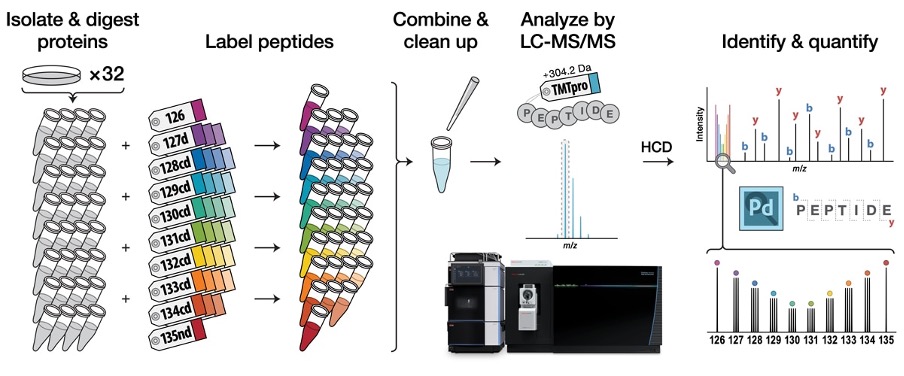
Tandem mass tags (TMT) are robust and efficient systems for multiplex, relative protein quantitation of up to 35 samples in a single LC-MS run. With bridged pooled standards, the TMT-based proteomics significantly boost the number of confident identifications and allow for better quantification between relative samples.
Targeted PRM Analysis
We use scheduled parallel reaction monitoring (PRM) to precisely measure proteins of interest in complex samples. This targeted approach allows us to focus on defined peptides with high sensitivity and reproducibility, making it well suited for biomarker studies. In our lab, we have developed PRM methods for the quantification of clinically relevant protein biomarkers, providing robust tools for monitoring disease-related changes and validating candidate biomarkers.
Absolute Quantitation using Spike-in Heavy Peptide Standard
Commercially available PQ500 Reference Peptides kit, containing 800 peptides from 500 proteins, are used for plasma proteomics. PQ500 offers absolute quantification of hundreds of human plasma proteins through the combination of the quantitative capabilities of MRM/PRM with the comprehensiveness of DIA
In the highly specialized lipoprotein research area, we have developed an in-house Apolipoprotein Heavy Peptide Standard panel for all 14 apolipoproteins with 44 different peptides, covering different isoforms/variants of Apo(a), ApoA1, ApoA2, APOA4, APOB, APOC1, APOC2, APOC3, APOD, APOE, APOH, APOL1, APOM, CLUS.
10.1038/s41467-021-23494-1
10.1161/CIRCRESAHA.122.321690
Post-Translational Modification
Mass Spectrometry instruments can be used for the identification of varies post translational modifications on proteins, such as phosphorylation, glycosylation and oxidation. Using our unique HCD-product dependent-ETD method and using Byonic software, we can identify the glycopeptide amino acid sequences, as well as the glycan site and composition of attached glycans, in a single MS/MS run. This especially useful for the extracellular matrix study on cardiovascular diseases.
10.1161/ATVBAHA.118.312175
10.1074/mcp.m112.024018
Cross-linking mass spectrometry
We use protein cross-linking mass spectrometry (XLMS) to study protein structures and interactions. By analysing these links with mass spectrometry and MS3 we can see how proteins are arranged and where they interact, allowing us to study even large and flexible protein complexes. XlinkX node in Proteome Discoverer is really useful for the protein identification and determination of cross-linked peptides.